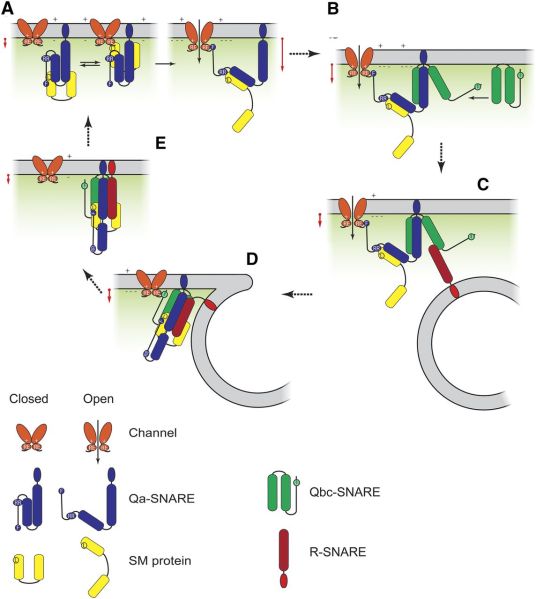
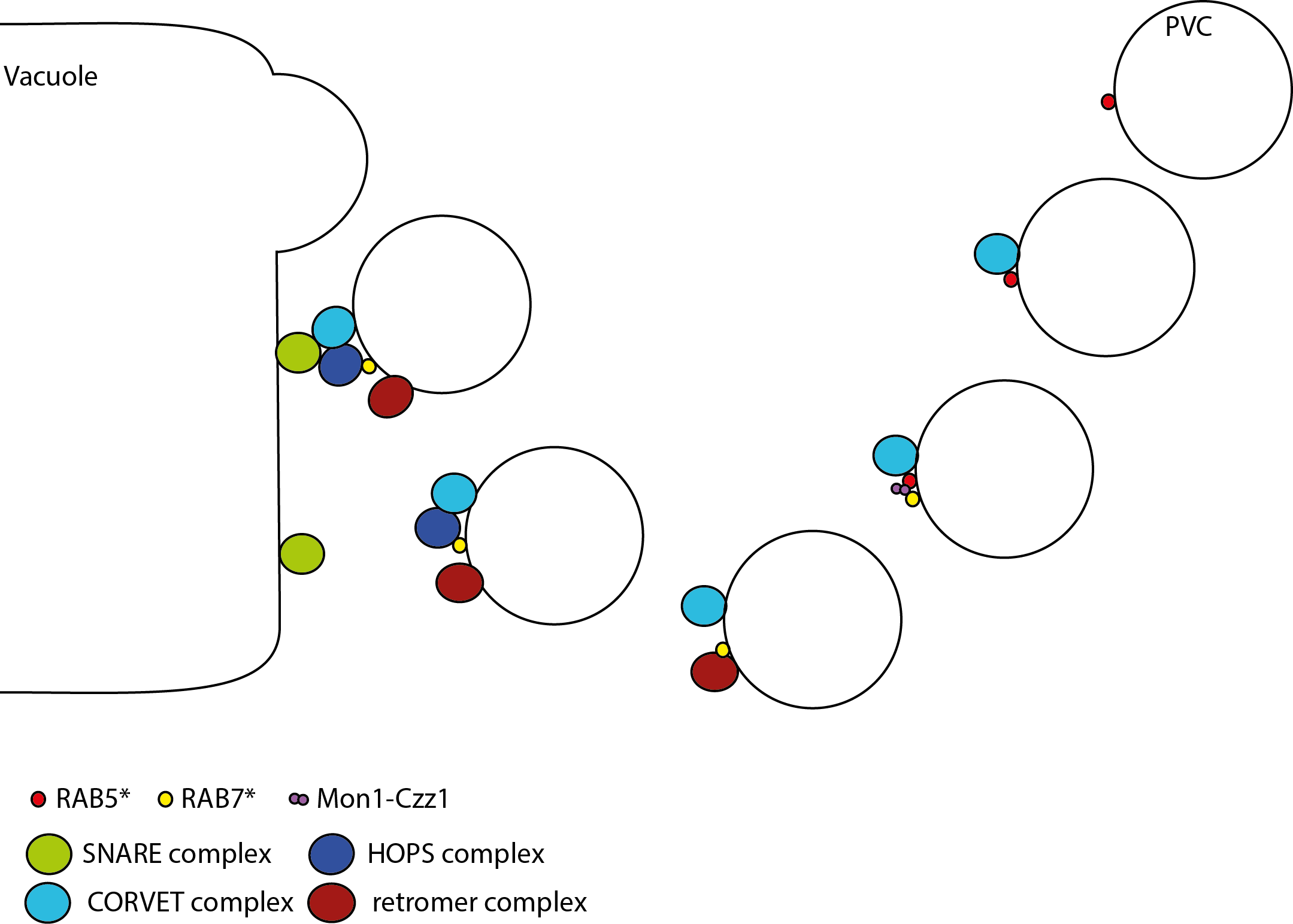
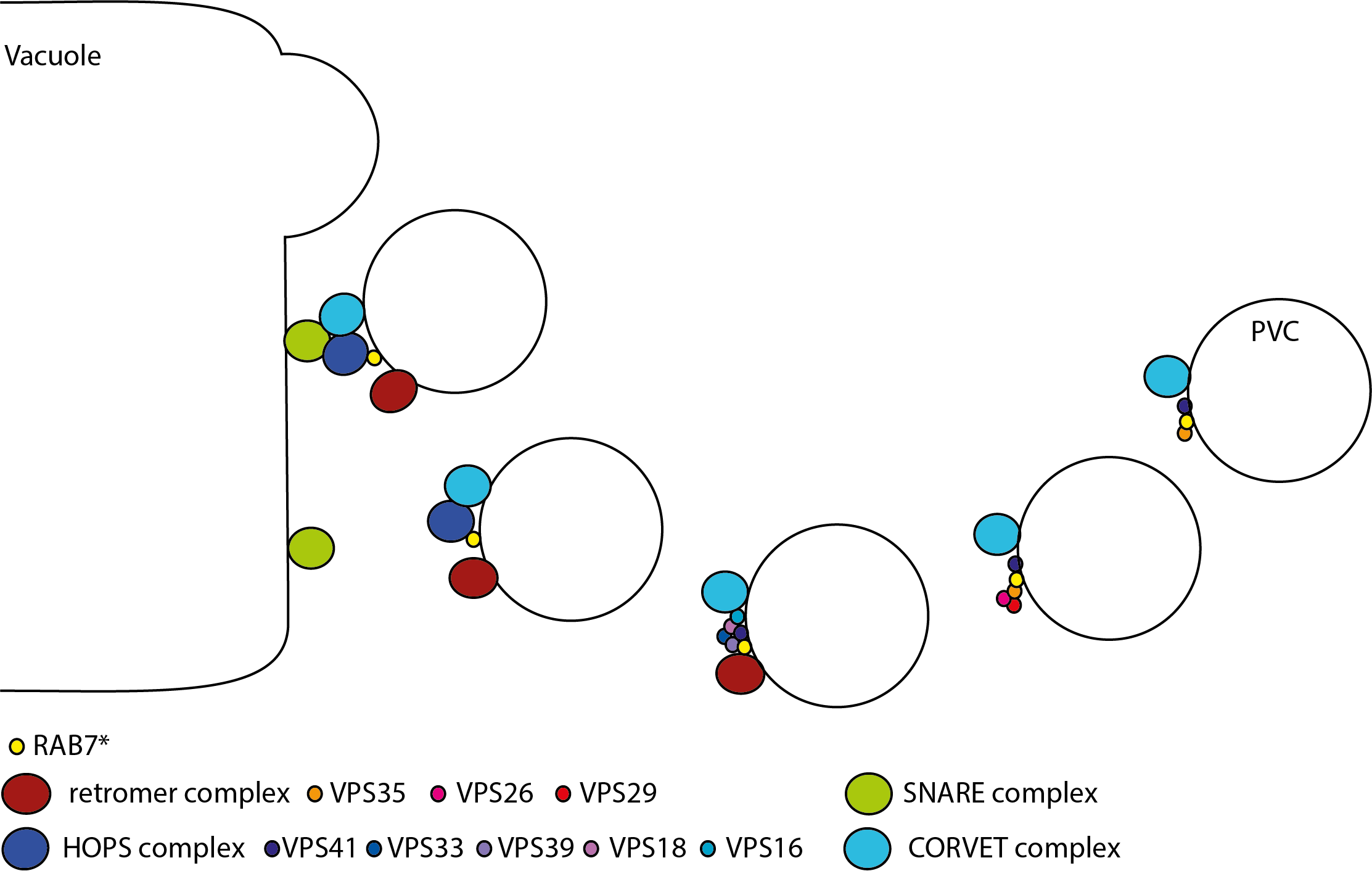
Giving a talk
When preparing to give a talk about your work you always need to make lots of decisions. One is about the amount of background vs results. Ideally you would like to have lots of time to discuss your new results, but for the audience to place them into context, or to understand them in the first place they will need some background. So they will know what you will be taking about. And here lies the difficulty. When you are talking about a widely known topic you might be able to spend a minute or two recapping what everybody already knows, before delving into your results and why they are so exciting. However, when you are studying a highly specialised field, where few people know the ins and outs, it might be wise to spent some more time bringing the audience up to speed. Yes this eats into valuable minutes that you might otherwise spend on talking excitingly about your latest research. But all this excitement will be for nothing if nobody else is understanding what you do. Of course, when you are in a highly specialised field and having an audience from that field you can bring you introduction back to a minute or two.
Giving a talk to a mixed audience with people that know the field well, e.g. your supervisor, and others that are doing completely different things, is tricky. Add to the fact that apparently not only am I speaking about one specialised field but two, phosphoinositides and a biochemistry approach, makes it that I some how lose the audience. Most of the time I forget that taking a biochemistry approach is less common than I think, having always done projects that uses biochemistry in one form or another. So I tend to focus on the new stuff for me, phosphoinositides and what they do. I will be giving these my larger share of background time. I don’t forget to talk through the methods that I used. But, I might forget to expand on why an approach is taken, assuming that it is obvious.
One of the things I apparently should have clarified a bit more in my latest talk, is on why comparing an enrichment in peripheral membrane proteins after salt stress treatment of only 30 minutes would tell us something about the proteins that interact with phosphoinositides under salt stress. The confusion was stemming from the fact that 30 minutes is not long enough for proteins that are synthesised in response to salt stress to be present. It simply takes longer to go through gene activation, transcription and protein synthesis.
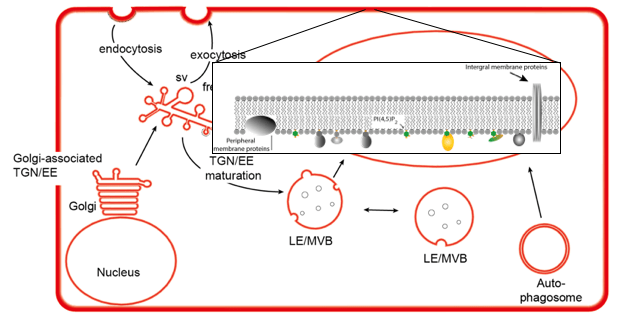
So it follows that the proteins interacting with phosphoinositides in response to 30 minutes of salt stress are already in the cytosol. In my talk I focussed on that the cytosol contains lots of proteins of which some will be able to interact with phosphoinositides. A lot of these interactions might be aspecific, so an enrichment for specific interactions would make sense. And that the proteins that interact with phosphoinositides are loosely attached to the membrane in the form of peripheral membrane proteins. Therefore, that it makes sense to use a protein extract enriched for peripheral membrane proteins in the interaction assay for identification of the phosphoinositide interactors. What I forgot to clarify was that the peripheral membrane proteins vary depending on the specifics of the cell. So will a non-stressed cell have different peripheral membrane proteins than a stressed cell. Salt stress and heat stress will attract different proteins to the membrane. Just as a cell in the root will have a different subset of peripheral membrane proteins than a cell in a leaf. Knowing this last bit of information and it makes complete sense to have a 30 minute salt stress treatment after which you compare your stressed with non-stressed samples. Not realising this and you might think that after 30 minutes of stress treatment you are still dealing with the same group of proteins as input for your interaction assay.
Now I just have to find a way to wave this into my next talk without it eating up to many precious minutes as I will have lots of exciting results to talk about as well.